
荧光共定位分析(colocalization),是一种重要的荧光分析方法。
它的本质其实就是分析不同荧光标记的蛋白(这两种荧光必须有独立的发射波长),在空间中的重叠,从而判断这两种蛋白是否处于同一区域,即在同一像素上是否“恰巧”出现了两种不同的荧光分子。
共定位分析能间接地说明两个蛋白与同一结构有联系,但不是两种蛋白有相互作用的直接证据。荧光偏振能量转移(FRET)是研究蛋白质之间相互作用的一种比较直观的方法。
例如发表在Science Signaling上的这篇文章就用到了荧光共定位分析[1]:
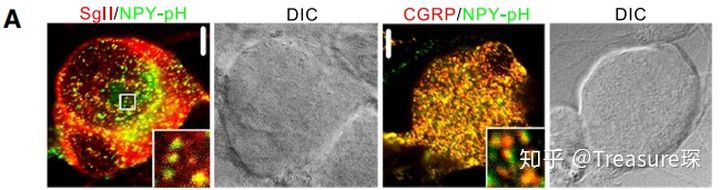

为了证明NPY-pH(用绿色荧光标记)这种蛋白被分选在了某种类型的囊泡中,对这种囊泡的两种marker——Sgll和CGRP进行染色(用红色荧光标记),然后进行共定位分析。如果绿色荧光和红色荧光“总是”同时出现,那可以说明NPY-pH这种蛋白确实被分选到了这种囊泡当中。
作为定量分析篇,这篇文章就为大家介绍怎样利用ImageJ进行荧光共定位分析。
首先,与往常不同,荧光共定位分析的操作十分简单。但实现这一分析的算法有很多种。
这些其实都是为了回答一个问题,即怎么定量描述共定位分析。现在最常用的有以下两种算法:
1、皮尔森相关系数——Pearson’s correlation coefficient (PCC)
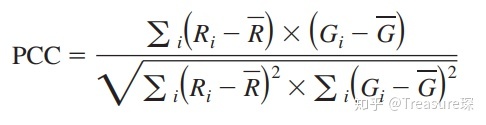
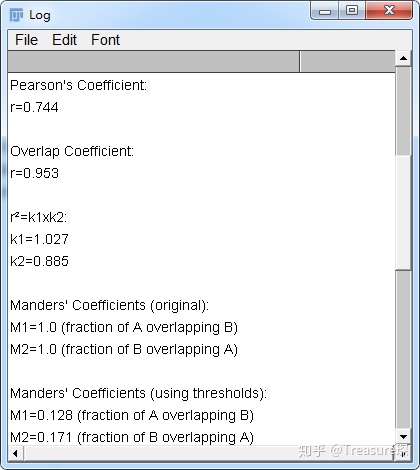
PCC是最常用的定量描述共定位的相关系数,取值在1到-1之间。1表示完美相关(有蛋白A的地方必有蛋白B); -1表示完全排除(有蛋白A地方必定没有蛋白B),零表示随机关系(蛋白A和蛋白B随机分布,无联系)。
2、曼德斯共定位系数——Manders’ Colocalization Coefficients (MCC)
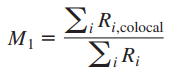
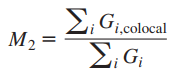
M1、M2代表一种蛋白质与另一种蛋白质共定位的部分,占这种蛋白总量的比例。相当于是重叠比例。
这两种种算法(其实还有更多算法)都集成在了插件中,它们各有各的适用范围和优缺点。
这篇文章会介绍三种不同的ImageJ插件来完成这一分析。但共定位分析真正重要的,或者说是真正需要我们去思考的,是它背后的原理。这些算法都不是完美的,不同情况下,利用不同算法的参数,才能得到准确的结果。
这一篇先讲操作,共定位分析原理的解析见下一片文章。
如果想正确的应用这一工具,请先阅读原理解析和ImageJ官网对于共定位分析的说明,以及一篇综述[2]:A practical guide to evaluating colocalization in biological microscopy。
Don’t treat this tool as a black box .
一、荧光共定位的操作(三种插件)
(1)Coloc 2(Analyze-Colocalization-Coloc 2)Fiji自带。下载地址:点击这里

这一插件是现在最新的共定位分析插件,而且说明也十分详细,集成了过去用于共定位分析的插件的很多功能。
这里我以ImageJ官网给出的一个样本为例,大家可以自行下载:点击这里
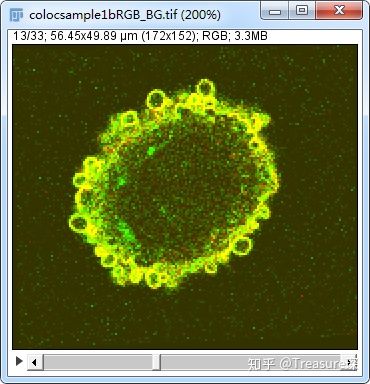
1、分离通道,保留有目标蛋白的两个通道(Image-Color-Split Channels)
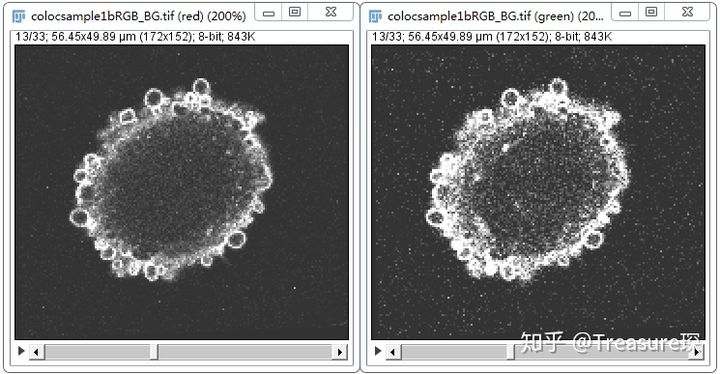
这时可以看到这两个通道是黑白,没有颜色的。
2、给两个通道分别添加伪色(Image-Color-Channels Tool)
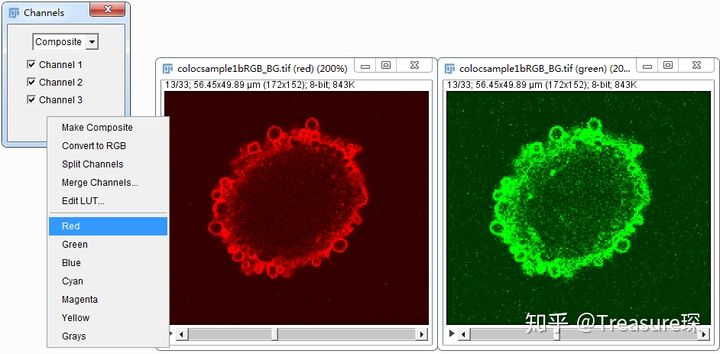
点击Channels界面里的More,给两个通道上色(Optional,只是为了更好区分两个通道)。
注意:Coloc2不能只能处理两张单张的图片,不能以Stack的形式进行分析,所以这里需要Split Channels,把这两张图片分别提取出来,进行后续的分析。
3、打开Colorc 2插件(Analyze-Colocalization-Coloc 2)
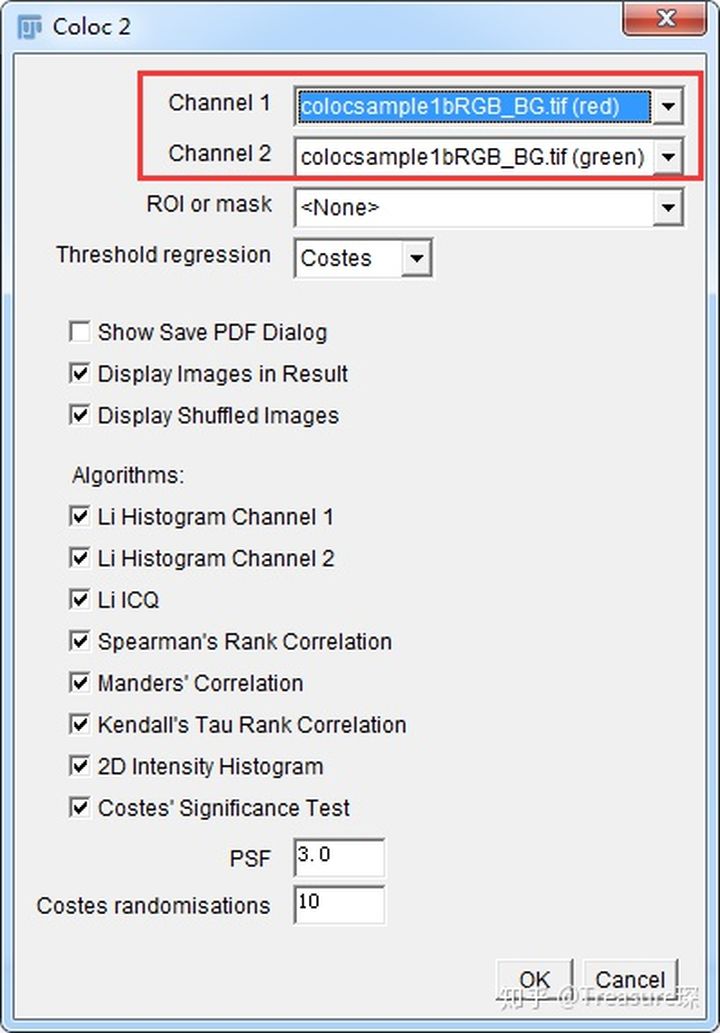
选好两个Channel,算法都勾选上,点击OK。然后会出现一个类似报告的结果,里面有皮尔森系数等等结果。
注意:图片如果为16-bit,应该提前转为8-bit,否则计算时间会非常长且结果有误。
在这里选择2D intensity histogram,即可得到散点图Scatter plot。
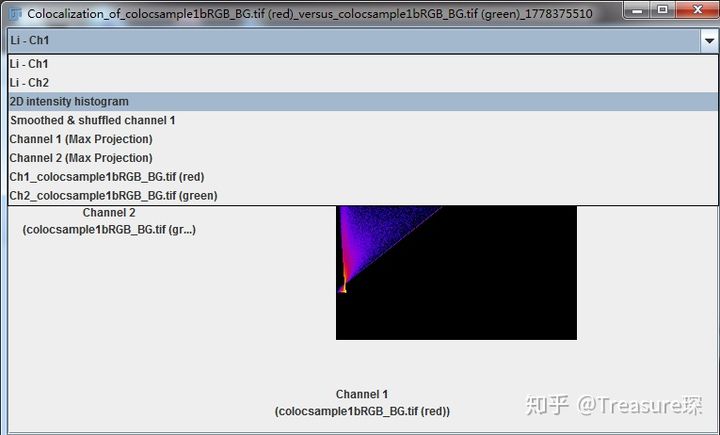
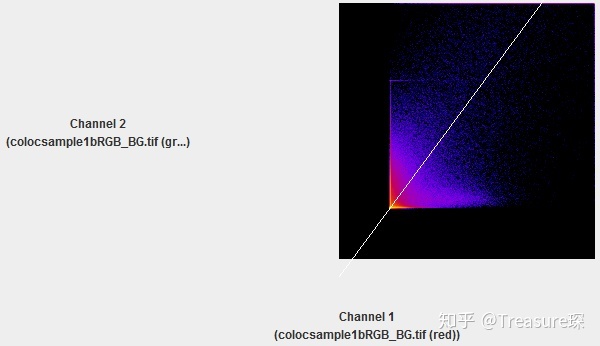
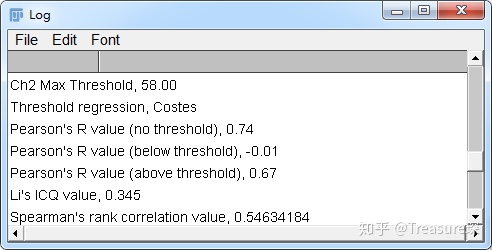
这些结果可以保存为pdf,也可以将数据导出。
注意:1、Threshold是指对图片的灰度设定一个阈值,高于多少才能被计算,作用是降低背景,去除不必要的。如果图片中物体与背景区分明显,可以很好进行Threshold,建议用above threshold的值。
2、建议将想要分析的区域框选出来,即选择特定的ROI,而不是对整张照片进行分析,具体原因可以参照解析。
(2)JACoP
WIKI:地址链接
下载地址:点击这里
这个插件需要在上面两个网址上下载,并放到plugins目录下,或者直接在Fiji的搜索框中搜索并安装。对于图像的预处理和前面一致。关于这个插件的原理和使用,可以参考这篇综述[3]:A guided tour into subcellular colocalization analysis in light microscopy.
3、完成上面的第1、2两步操作后,打开JACoP插件(Plugins-JACoP)
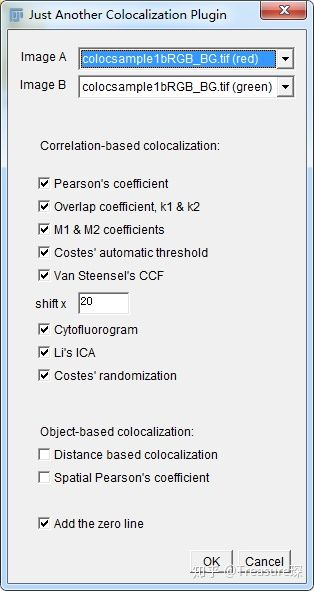
勾选上所有的算法,然后点击OK。
这里填拍这张照片用的显微镜的信息,以及荧光的发射波长。
默认设置,点击OK。
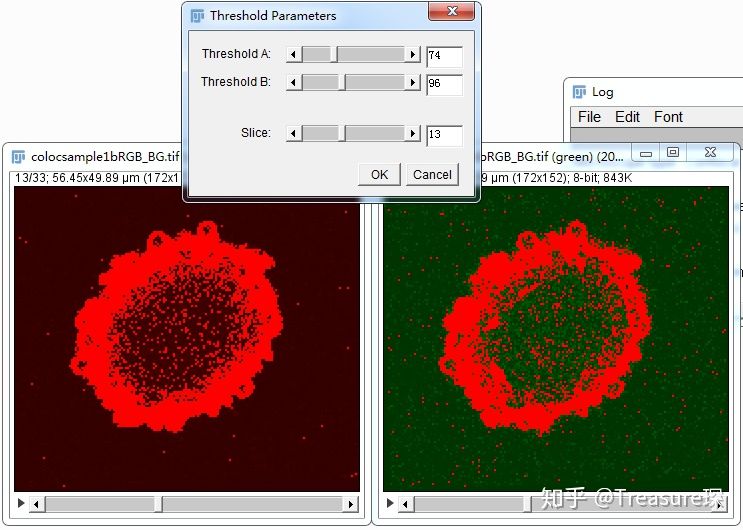
设定适当的阈值,尽量减少背景,包含感兴趣的区域,点击OK即可得到结果。
(3)Colocalization Finder
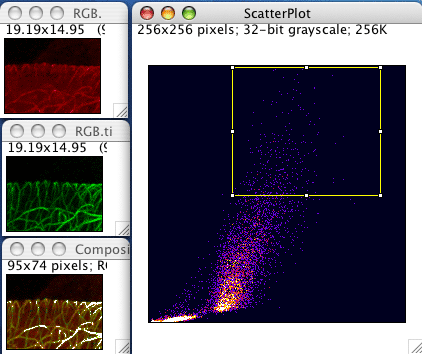
这个插件需要在这里下载(点击这里),并放到plugins目录下,或者直接在Fiji的搜索框中搜索并安装。对于图像的预处理和前面一致。
这个插件现在已经没有人进行开发了,所以官网上建议使用最新的Coloc 2插件。
3、完成上面的第1、2两步操作后,打开Colocalization Finder插件(Plugins-Colocalization Finder)
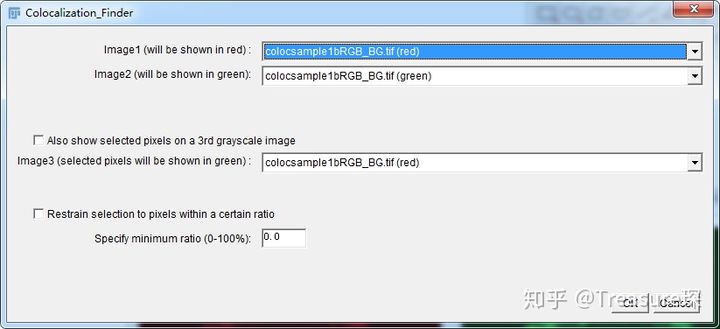
选中两个Channel后点击OK,即可得到结果。
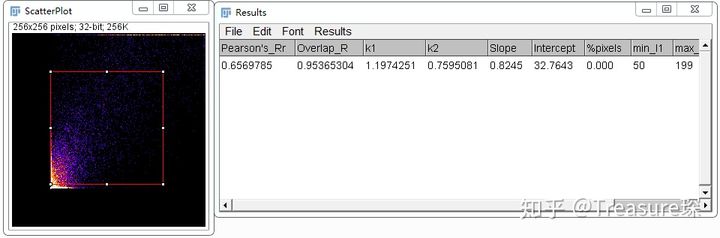
可见这个插件计算出的结果,和前两个插件有一定差别,建议使用前两款插件。
二、Plot Profile进行共定位分析描述
网上其实也有利用Plot Profile进行共定位分析,但这种方法其实只能描述,并不准确。
我以ImageJ自带的样本(File-Open Samples-Fluorescent Cells)为例,具体操作如下:
1、打开图片后,用直线/矩形工具框选感兴趣区域
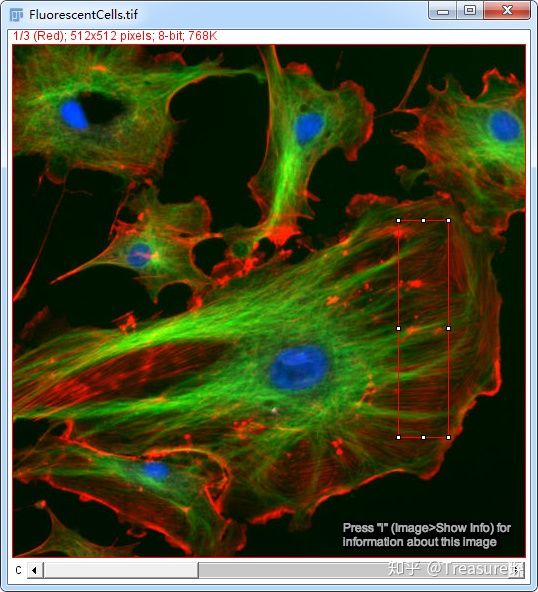
注意左上角,这时候其实是红色这一通道。
2、打开Plot Profile(Analyze-Plot Profile)
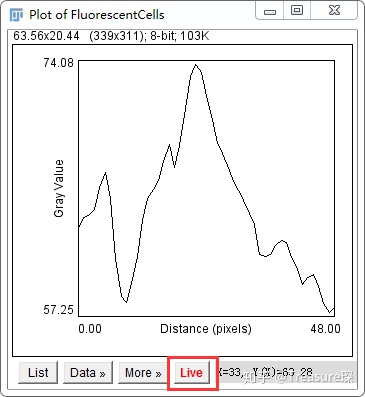
这是红色通道荧光强度(灰度)沿着矩形的分布,然后点击live。
这时候向右拉到绿色通道,Plot Profile就会实时变化:
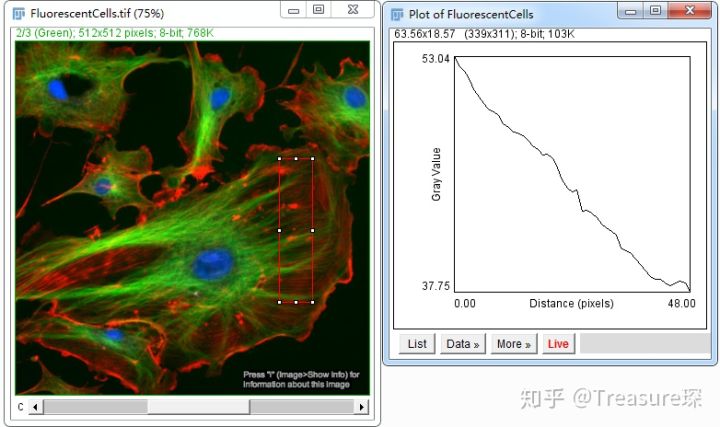
分别将Data导出,然后在同一坐标系中作图,即可得到不同荧光沿着该矩形距离的变化曲线。
为什么一个看似简单的共定位分析,需要用这么多方法、这么多参数来描述?为什么需要了解其中的原理?
下一篇我会对这些问题进行解答,敬请期待。
希望对大家有帮助~
参考文献:
[1].Wang Y , Wu Q , Hu M , et al. Ligand- and voltage-gated Ca2+ channels differentially regulate the mode of vesicular neuropeptide release in mammalian sensory neurons[J]. Science Signaling, 2017, 10(484).
[2].Dunn K W , Kamocka M M , Mcdonald J H . A practical guide to evaluating colocalization in biological microscopy[J]. AJP: Cell Physiology, 2011, 300(4):C723-C742.
[3].Bolte S , F. P. CORDELIèRES. A guided tour into subcellular colocalization analysis in light microscopy[J]. Journal of Microscopy (Oxford), 2006, 224(3):213-232.
